
BRUKINSA 80 mg, gélule, boîte de 1 flacon de 120
Dernière révision : 07/05/2025
Taux de TVA : 2.1%
Prix de vente : 4 750,87 €
Taux remboursement SS : 100%
Base remboursement SS : 4 750,87 €
Laboratoire exploitant : BEIGENE FRANCE
Source :

BRUKINSA en monothérapie est indiqué pour le traitement de patients adultes atteints de macroglobulinémie de Waldenström (MW) qui ont reçu au moins un traitement antérieur, ou pour le traitement en première intention de patients inéligibles à une chimio-immunothérapie.
BRUKINSA en monothérapie est indiqué pour le traitement de patients adultes atteints de lymphome de la zone marginale (LZM) ayant reçu au moins un traitement antérieur à base d'anticorps anti-CD20.
BRUKINSA en monothérapie est indiqué pour le traitement de patients adultes atteints de leucémie lymphoïde chronique (LLC).
BRUKINSA en association avec l'obinutuzumab est indiqué pour le traitement de patients adultes atteints de lymphome folliculaire (FL) réfractaire ou en rechute qui ont reçu au moins deux traitements systémiques antérieurs.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hémorragie
Des événements hémorragiques graves et fatals ont été observés chez des patients traités par BRUKINSA. Des événements de saignement de grade 3 ou plus, notamment hémorragie intracrânienne et gastro-intestinale, hématurie et hémothorax, ont été signalés chez des patients (voir rubrique Effets indésirables). Des événements de saignement de tout grade, notamment purpura et pétéchies, sont survenus chez des patients atteints de tumeurs malignes hématologiques. Le mécanisme des évènements de saignement n'est pas bien compris.
BRUKINSA peut augmenter le risque d'hémorragie chez les patients sous traitement antiplaquettaire ou anticoagulant, et les patients doivent faire l'objet d'une surveillance pour rechercher des signes de saignement. Une modification de la dose peut s'avérer nécessaire en cas d'effet indésirable de grade 3 ou supérieur, conformément aux recommandations (voir rubrique Posologie et mode d'administration). La warfarine ou d'autres antagonistes de la vitamine K ne doivent pas être administrés en même temps que BRUKINSA. Il est nécessaire de surveiller les signes et symptômes de saignement et la numération formule sanguine des patients. Il convient d'évaluer les risques et les bénéfices d'un traitement anticoagulant ou antiplaquettaire en cas d'administration concomitante avec BRUKINSA. En fonction du type d'intervention chirurgicale et du risque de saignement, il convient d'évaluer le rapport bénéfice/risque de la suspension du zanubrutinib pendant 3 à 7 jours avant et après l'intervention chirurgicale.
Infections
Des infections fatales ou non (notamment infections bactériennes, virales, fongiques ou sepsis) ainsi que des infections opportunistes (par ex., des infections virales herpétiques, cryptococciques, à Aspergillus et à Pneumocystis jiroveci) ont été observées chez des patients traités par BRUKINSA. Des infections de grade 3 ou plus sont survenues chez des patients (voir rubrique Effets indésirables). L'infection de grade 3 ou plus la plus fréquente était la pneumonie. Des infections dues à une réactivation du virus de l'hépatite B (VHB) se sont également produites. Avant d'initier le traitement par BRUKINSA, le statut du VHB des patients doit être établi. Une consultation chez un médecin expert en maladie hépatique est recommandée pour les patients dont le test du VHB est positif ou dont la sérologie pour l'hépatite B est positive, avant d'initier le traitement. Il convient de surveiller et prendre en charge les patients, conformément aux traitements recommandés, afin de prévenir une réactivation de l'hépatite B. Envisager une prophylaxie par le traitement standard chez les patients présentant un risque accru d'infections. Les patients doivent être surveillés pour détecter d'éventuels signes et symptômes de l'infection et les traiter de façon appropriée.
Cytopénies
Des cytopénies de grade 3 ou 4, notamment neutropénie, thrombopénie et anémie, identifiées par des mesures biologiques ont été signalées chez des patients traités par BRUKINSA (voir rubrique Effets indésirables). Surveiller la formule sanguine complète pendant le traitement (voir rubrique Posologie et mode d'administration).
Secondes tumeurs malignes primitives
Des secondes tumeurs malignes primitives, notamment carcinome non cutané, ont été observées chez des patients atteints de tumeurs malignes hématologiques traités par BRUKINSA. La seconde tumeur maligne primitive la plus fréquente était le cancer de la peau (carcinome basocellulaire et épidermoïde de la peau). Conseiller aux patients d'utiliser une protection solaire.
Fibrillation et flutter auriculaire
Des fibrillations et des flutters auriculaires ont été observés chez des patients atteints de tumeurs malignes hématologiques traités par BRUKINSA, en particulier chez des patients présentant des facteurs de risque cardiaque, une hypertension, des infections aiguës et chez les personnes âgées (≥ 65 ans). Surveiller les signes et symptômes de fibrillation et de flutter auriculaires et les prendre en charge comme il convient.
Syndrome de lyse tumorale
Le syndrome de lyse tumorale a été rarement rapporté avec le traitement par zanubrutinib en monothérapie, en particulier chez les patients traités pour une leucémie lymphoïde chronique (LLC). Évaluer les risques pertinents (par exemple, un fardeau tumoral élevé ou taux élevé d'acide urique sanguin) et prendre les précautions appropriées. Surveiller attentivement les patients et traiter le cas échéant.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode de contraception hautement efficace pendant le traitement par Brukinsa (voir rubrique Fertilité, grossesse et allaitement).
BRUKINSA contient du sodium.
BRUKINSA contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Zanubrutinib en monothérapie
Les effets indésirables les plus fréquents (≥ 20 %) du zanubrutinib en monothérapie sont une infection des voies aériennes supérieures§ (36 %), des ecchymoses§ (32 %), une hémorragie/des hématomes (30 %), la neutropénie§ (30 %), des douleurs musculosquelettiques§ (27 %), un rash§ (25 %), une pneumonie§ (24 %), de la diarrhée (21 %) et de la toux§ (21 %) (Tableau 3).
Les effets indésirables de grade 3 ou plus les plus fréquents (> 3 %) du zanubrutinib en monothérapie sont la neutropénie§ (21 %), la pneumonie§ (14 %), l'hypertension (8 %), la thrombopénie§ (6 %), l'anémie (6 %) et l'hémorragie/les hématomes§ (4 %).
Parmi les 1 550 patients traités par zanubrutinib, 4,8 % des patients ont arrêté le traitement en raison d'effets indésirables. L'effet indésirable le plus fréquent ayant entraîné un arrêt du traitement était la pneumonie§ (2,6 %). 5,0 % des patients ont eu un effet indésirable ayant entraîné une réduction de la dose.
Zanubrutinib en association avec l'obinutuzumab
Les effets indésirables les plus fréquents (≥ 20 %) du zanubrutinib en association avec l'obinutuzumab ont été la thrombocytopénie§ (37 %), la neutropénie§ (31 %) et la fatigue§ (27 %) (Tableau 4).
Les effets indésirables de grade 3 ou plus les plus fréquents (> 3 %) du zanubrutinib en association avec l'obinutuzumab ont été la neutropénie§ (25 %), la thrombocytopénie§ (16 %), la pneumonie§ (15 %) et l'anémie (5 %).
Sur les 143 patients traités par zanubrutinib en association avec l'obinutuzumab, 4,9 % ont arrêté le traitement en raison d'effets indésirables. L'effet indésirable le plus fréquent ayant conduit à l'arrêt du traitement était la pneumonie§ (4,2 %). Des effets indésirables ayant entraîné une réduction de la dose sont survenus chez 7,0 % des patients.
Une diminution de la numération plaquettaire† (basée sur les valeurs biologiques) a été observée chez 65 % (tous grades confondus) et 12 % (grade 3 ou 4) des patients recevant le zanubrutinib en association avec l'obinutuzumab, contre 43 % (tous grades confondus) et 11 % (grade 3 ou 4) chez les patients recevant l'obinutuzumab. Une diminution de la numération plaquettaire de tous grades et de grade 3 ou 4 a été rapportée chez 39 % et 7,8 % des patients ayant reçu le zanubrutinib en monothérapie.
Tableau récapitulatif des effets indésirables
Le profil de sécurité repose sur les données regroupées de 1 550 patients atteints de tumeurs malignes impliquant les lymphocytes B, y compris des patients atteints de leucémie lymphoïde chronique (N = 938), de macroglobulinémie de Waldenström (N = 249), de lymphome à cellules du manteau (N = 140), de lymphome de la zone marginale (N = 93), de lymphome folliculaire (N = 59) et d'autres types de tumeurs malignes impliquant les lymphocytes B (N= 71), traités par BRUKINSA au cours d'études cliniques avec une durée médiane d'exposition de 34,41 mois.
Le profil de sécurité du zanubrutinib en association avec l'obinutuzumab est basé sur les données de l'étude ROSEWOOD portant sur 143 patients atteints de lymphome folliculaire et traités par BRUKINSA en association avec l'obinutuzumab, avec une durée médiane d'exposition de 12,35 mois.
Les effets indésirables observés chez les patients traités par BRUKINSA en monothérapie ou en association avec l'obinutuzumab pour des tumeurs malignes impliquant les lymphocytes B sont énumérés ci-dessous au Tableau 3 et au Tableau 4, respectivement, par classe de système d'organes et groupe de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de sévérité.
Tableau 3 : effets indésirables du zanubrutinib en monothérapie signalés dans les études cliniques menées chez des patients atteints de tumeurs malignes impliquant les lymphocytes B (n = 1550)
|
SOC
selon le dictionnaire MedDRA |
Termes
selon le dictionnaire MedDRA |
Tous
grades* (%) |
Grade
3 ou plus (%) |
|
Infections
et infestations |
Infection
des voies aériennes supérieures§ |
Très fréquent (36) | 2 |
| Pneumonie§# | Très fréquent (24) | 14 | |
| Pneumonie | Très fréquent (15) | 8 | |
| Infection des voies aériennes inférieures | Fréquent (5) | < 1 | |
| Infection des voies urinaires | Très fréquent (14) | 2 | |
| Bronchite | Fréquent (4) | < 1 | |
| Réactivation de l'hépatite B | Peu fréquent (< 1) | < 1 | |
|
Affections
hématologiques et du système lymphatique |
Neutropénie§ | Très fréquent (30) | 21 |
| Neutropénie fébrile | Fréquent (2) | 2 | |
| Thrombopénie§ | Très fréquent (18) | 6 | |
| Anémie§ | Très fréquent (16) | 6 | |
|
Affections
du système nerveux |
Vertiges§ | Très fréquent (12) | < 1 |
|
Affections
cardiaques |
Fibrillation et flutter atriaux | Fréquent (5) | 2 |
|
Affections
vasculaires |
Ecchymoses§ | Très fréquent (32) | < 1 |
| Contusion | Très fréquent (20) | 0 | |
| Pétéchies | Fréquent (7) | < 1 | |
| Purpura | Fréquent (5) | < 1 | |
| Ecchymose | Fréquent (3) | < 1 | |
| Hémorragie/hématome§# | Très fréquent (30) | 3 | |
| Hématurie | Très fréquent (11) | < 1 | |
| Épistaxis | Fréquent (8) | < 1 | |
| Hémorragie gastro-intestinale | Peu fréquent (< 1) | < 1 | |
| Hypertension§ | Très fréquent (17) | 8 | |
|
Affections
gastro- intestinales |
Diarrhée | Très fréquent (21) | 2 |
| Constipation | Très fréquent (14) | < 1 | |
|
Affections
de la peau et du tissu sous-cutané |
Rash§ | Très fréquent (25) | < 1 |
| Prurit | Fréquent (8) | < 1 | |
| Dermatite exfoliative généralisée |
Fréquence
indéterminée |
Fréquence
indéterminée |
|
|
Affections
musculo- squelettiques et systémiques |
Douleur musculo-squelettique§ | Très fréquent (27) | 2 |
| Arthralgie | Très fréquent (15) | < 1 | |
| Dorsalgie | Très fréquent (12) | < 1 | |
|
Troubles
généraux et anomalies au site d'administration |
Fatigue§ | Très fréquent (18) | 1 |
| Fatigue | Très fréquent (14) | 1 | |
| Asthénie | Fréquent (4) | < 1 | |
| Œdème périphérique | Fréquent (9) | < 1 | |
|
Affections
respiratoires, thoraciques et médiastinales |
Toux§ | Très fréquent (21) | < 1 |
|
Troubles
du métabolisme et de la nutrition |
Syndrome de lyse tumorale§# | Peu fréquent (< 1) | < 1 |
| Investigations† | Réduction du nombre de neutrophiles†± | Très fréquent (52) | 22 |
| Réduction du nombre de plaquettes†± | Très fréquent (39) | 8 | |
| Réduction de l'hémoglobine†± | Très fréquent (26) | 4 |
* Les
grades ont été évalués selon les critères communs de terminologie pour les
événements indésirables de l'Institut national américain du cancer (NCI-CTCAE),
version 4.03.
† Basé sur des mesures biologiques.
± Les pourcentages sont basés sur le nombre de patients disposant
d'évaluations à la référence et au moins une fois ultérieurement.
§ Comprend plusieurs termes d'effets indésirables
# Comprend les événements d'issue fatale
Tableau 4 : effets indésirables du zanubrutinib en association avec l'obinutuzumab rapportés dans l'étude ROSEWOOD (BGB-3111-212) menée chez des patients atteints de lymphome folliculaire (n = 143)
|
SOC
selon le dictionnaire MedDRA |
Termes
selon le dictionnaire MedDRA |
||
|
Tous
grades* (%) |
Grade
3 ou plus (%) |
||
| Infections et infestations |
Infection
des voies aériennes supérieures§ |
Très fréquent (14) | < 1 |
| Pneumonie§# | Très fréquent (20) | 15 | |
| Pneumonie | Très fréquent (13) | 11 | |
| Infection
des voies aériennes inférieures |
Fréquent (4) | < 1 | |
| Infection des voies urinaires | Fréquent (10) | 2 | |
| Bronchite | Fréquent (2) | 0 | |
|
Affections hématologiques et du système lymphatique |
Thrombopénie§ | Très fréquent (37) | 16 |
| Neutropénie§ | Très fréquent (31) | 25 | |
| Anémie§ | Très fréquent (12) | 5 | |
|
Affections
du système nerveux |
Vertiges§ | Fréquent (4) | 0 |
| Affections cardiaques | Fibrillation auriculaire et flutter§ | Fréquent (3) | 1 |
| Affections vasculaires | Hémorragie/hématome§ | Très fréquent (16) | < 1 |
| Épistaxis | Fréquent (5) | 0 | |
| Hématurie | Fréquent (< 1) | 0 | |
| Ecchymoses§ | Très fréquent (15) | 0 | |
| Contusion | Très fréquent (8) | 0 | |
| Pétéchies | Fréquent (6) | 0 | |
| Purpura | Fréquent (2) | 0 | |
| Ecchymose | Fréquent (1) | 0 | |
| Hypertension§ | Fréquent (4) | < 1 | |
|
Affections
gastro- intestinales |
Diarrhée | Très fréquent (19) | 3 |
| Constipation | Très fréquent (13) | 0 | |
|
Affections
de la peau et du tissu sous-cutané |
Rash§ | Très fréquent (10) | 0 |
| Prurit | Fréquent (7) | 0 | |
| Dermatite exfoliative généralisée |
Fréquence indéterminée |
Fréquence
indéterminée |
|
|
Affections
musculo- squelettiques et systémiques |
Douleur musculo-squelettique§ | Très fréquent (18) | 2 |
| Dorsalgie | Très fréquent (11) | < 1 | |
| Arthralgie | Fréquent (4) | 0 | |
|
Troubles
généraux et anomalies au site d'administration |
Fatigue§ | Très fréquent (27) | 1 |
| Fatigue | Très fréquent (15) | 0 | |
| Asthénie | Fréquent (12) | < 1 | |
| Œdème périphérique | Fréquent (2) | 0 | |
|
Affections
respiratoires, thoraciques et médiastinales |
Toux§ | Très fréquent (13) | 0 |
|
Investigations†
|
Diminution du nombre de plaquettes†± | Très fréquent (65) | 12 |
| Diminution du nombre absolu de neutrophiles†± | Très fréquent (48) | 18 | |
| Diminution de l'hémoglobine†± | Très fréquent (31) | < 1 | |
* Les grades ont été évalués selon les critères communs de terminologie pour les événements indésirables de l'Institut national américain du cancer (NCI-CTCAE, version 5.0).
† Basé sur des mesures biologiques.
§ Comprend plusieurs termes d'effets indésirables
# Comprend les événements d'issue fatale
± Les pourcentages sont basés sur le nombre de patients disposant d'évaluations à la référence et au moins une fois ultérieurement.
Autres populations particulières
Personnes âgées
Parmi les 1 550 patients traités par BRUKINSA en monothérapie, 61,3 % étaient âgés de 65 ans ou plus. L'incidence des effets indésirables de grade 3 ou plus était légèrement plus élevée chez les patients âgés traités par zanubrutinib (69,6 % des patients âgés de ≥ 65 ans par rapport à 62,7 % des patients âgés de < 65 ans). Aucune différence cliniquement significative concernant la sécurité n'a été observée entre les patients de ≥ 65 ans et les patients plus jeunes.
Parmi les 143 patients traités par BRUKINSA en association avec l'obinutuzumab, 42,0 % étaient âgés de 65 ans ou plus. L'incidence des effets indésirables de grade 3 ou plus était légèrement plus élevée chez les patients âgés traités par le zanubrutinib en association avec l'obinutuzumab (70,0 % des patients âgés de ≥ 65 ans contre 62,7 % des patients âgés de < 65 ans). Aucune différence cliniquement pertinente en termes de sécurité d'emploi n'a été observée entre les patients âgés de ≥ 65 ans et les patients plus jeunes.
Population pédiatrique
La sécurité et l'efficacité de BRUKINSA chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT L'INITIATION DU TRAITEMENT :
- Etablir le statut du VHB des patients. En cas de test VHB
positif, conseiller au patient de consulter un médecin expert en
maladie hépatique . Il convient de surveiller et prendre en charge les
patients, conformément aux traitements recommandés, afin de prévenir
une réactivation de l'héatite B. Envisager une prophylaxie par le
traitement standard chez les patients présentant un risque accru
d'infections.
SURVEILLANCE DU TRAITEMENT:
- Les signes et symptômes de saignement.
- La formule sanguine complète.
- D'éventuels signes et symptômes d'infection.
CONSEILLER l'utilisation d'une protection solaire.
ARRETER LE TRAITEMENT ET CONSULTER IMMEDIATEMENT UN MEDECIN en cas d'éruption cutanée prurigineuse en relief, difficultés à respirer, gonflement du visage, des lèvres, de la langue ou de la gorge.
PREVENIR LE MEDECIN en cas de :
- fièvre, frissons, douleurs corporelles, sensation de fatigue, symptômes de rhume ou de grippe, essoufflement, miction fréquente et douloureuse ;
- ecchymoses ou tendance accrue à développer des ecchymoses, contusions ;
- saignement
- douleurs musculaires et osseuses ;
- diarrhée ;
- présence de sang dans les urines, sang dans les selles ;
- petites tâches de saignement sous la peau ;
- fréquence cardiaque rapide, battements cardiaques irréguliers, pouls faible ou irrégulier, étourdissements, essoufflement, gène thoracique ;
- mains, chevilles ou pieds enflés.
UTILISER une protection solaire.
EVITER LA PRISE de produits à base de plantes contenant du millepertuis (Hypericum perforatum).
PRUDENCE en cas de consommation de pamplemousse ou d'orange amère (orange de Séville).
PRUDENCE en cas de conduite de véhicules ou utilisation de machines (possibilité de fatigue, vertiges et asthénie).
FEMME EN AGE DE PROCREER : utiliser une méthode de contraception hautement efficace pendant le traitement et jusqu'à 1 mois après l'arrêt du traitement.
Femmes en âge de procréer/Contraception chez les femmes
D'après les résultats observés chez les animaux, BRUKINSA peut être nocif pour le fœtus lorsqu'il est administré chez la femme enceinte (voir rubrique Données de sécurité préclinique). Les femmes doivent éviter de démarrer une grossesse pendant le traitement par BRUKINSA et pendant jusqu'à 1 mois après l'arrêt du traitement. Par conséquent, les femmes en âge de procréer doivent utiliser des méthodes de contraception hautement efficaces pendant le traitement par BRUKINSA et jusqu'à 1 mois après l'arrêt du traitement. On ne sait pas actuellement si le zanubrutinib peut ou non diminuer l'efficacité des contraceptifs hormonaux ; par conséquent, les femmes utilisant une contraception hormonale doivent recourir en complément à une méthode de contraception mécanique. Un test de grossesse est recommandé pour les femmes en âge de procréer avant d'instaurer un traitement.
Grossesse
BRUKINSA ne doit pas être utilisé pendant la grossesse. Il n'existe pas de données sur l'utilisation de BRUKINSA chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Allaitement
On ne sait pas si le zanubrutinib ou ses métabolites sont excrétés dans le lait maternel, aucune étude non clinique n'a été réalisée. On ne peut exclure le risque pour l'enfant allaité. L'allaitement doit être arrêté pendant le traitement par Brukinsa.
Fertilité
Aucun effet sur la fertilité de rats mâles ou femelles n'a été observé, mais des anomalies morphologiques dans les spermatozoïdes et une augmentation des pertes après implantation ont été constatées à 300 mg/kg/jour (voir rubrique Données de sécurité préclinique).
Le zanubrutinib est principalement métabolisé par l'enzyme 3A du cytochrome P450 (CYP3A).
Agents pouvant augmenter les concentrations plasmatiques de zanubrutinib
L'utilisation concomitante de BRUKINSA et de médicaments qui inhibent fortement ou modérément CYP3A peut accroître l'exposition au zanubrutnib.
Inhibiteurs puissants du CYP3A
La co-administration de plusieurs doses d'itraconazole (inhibiteur puissant du CYP3A) chez des volontaires sains a multiplié la Cmax du zanubrutinib par 2,6 et son ASC par 3,8. La co-administration de plusieurs doses de voriconazole et de clarithromycine (inhibiteurs puissants du CYP3A) chez des patients atteints de tumeurs malignes impliquant des lymphocytes B a multiplié l'exposition au zanubrutinib par 3,30 et son ASC0-24h normalisée par la dose par 1,92. La Cmax normalisée par la dose a été multipliée par 3,29 et 2,01, respectivement.
Si un inhibiteur puissant du CYP3A doit être utilisé (par exemple, posaconazole, voriconazole, kétoconazole, itraconazole, clarithromycine, indinavir, lopinavir, ritonavir, télaprévir), il convient de réduire la dose de BRUKINSA à 80 mg (une gélule) pendant la durée de l'utilisation de l'inhibiteur. Le patient doit être étroitement surveillé quant à la toxicité et respecter les directives de modification de la dose si nécessaire (voir rubrique Posologie et mode d'administration).
Inhibiteurs modérés du CYP3A
La co-administration de plusieurs doses de fluconazole et de diltiazem (inhibiteurs modérés du CYP3A) chez des patients atteints de tumeurs malignes impliquant les lymphocytes B a multiplié l'exposition au zanubrutinib par 1,88 et son AUC0-24h par 1,62. La Cmax normalisée par la dose a été multipliée par 1,81 et 1,62, respectivement.
Si un inhibiteur modéré du CYP3A doit être utilisé (par exemple, érythromycine, ciprofloxacine, diltiazem, dronédarone, fluconazole, vérapamil, aprépitant, imatinib, jus de pamplemousse, oranges amères), il convient de réduire la dose de BRUKINSA à 160 mg (deux gélules) pendant la durée de l'utilisation de l'inhibiteur. Les patients doivent être soigneusement surveillés quant à la toxicité et suivre les directives de modification de la dose si nécessaire (voir rubrique Posologie et mode d'administration).
Inhibiteurs légers du CYP3A
Les simulations utilisant le jeûne ont suggéré que les inhibiteurs légers du CYP3A (par exemple, ciclosporine et fluvoxamine) peuvent augmenter l'ASC du zanubrutinib de < 1,5 fois. L'association avec des inhibiteurs légers n'exige aucun ajustement de dose. Les patients doivent être soigneusement surveillés quant à la toxicité et suivre les directives de modification de la dose si nécessaire.
Le pamplemousse et les oranges amères doivent être consommés avec prudence pendant le traitement par BRUKINSA car ils contiennent des inhibiteurs modérés du CYP3A (voir rubrique Posologie et mode d'administration).
Agents pouvant diminuer les concentrations plasmatiques de zanubrutinib
L'utilisation concomitante de zanubrutinib et d'inducteurs puissants ou modérés du CYP3A peut diminuer les concentrations plasmatiques de zanubrutinib.
Inducteurs du CYP3A
La co-administration de plusieurs doses de rifampine (inducteur puissant du CYP3A) a diminué de 92 % la Cmax de zanubrutinib et l'ASC de 93 % chez des sujets sains. L'utilisation concomitante d'inducteurs puissants du CYP3A (par exemple, carbamazépine, phénytoïne, rifampine, millepertuis) et d'inducteurs modérés du CYP3A (par exemple, bosentan, éfavirenz, étravirine, modafinil, nafcilline) doit être évitée (voir rubrique Posologie et mode d'administration). La co-administration de plusieurs doses de rifabutine (inducteur modéré du CYP3A) a diminué de 48 % la Cmax de zanubrutinib et l'ASC de 44 % chez des sujets sains. Les inducteurs légers du CYP3A peuvent être utilisés avec prudence pendant le traitement par BRUKINSA.
Agents anti-acide gastrique
Aucune différence cliniquement significative de la pharmacocinétique du zanubrutinib n'a été observée lors de la co-administration avec des agents réduisant l'acidité gastrique (inhibiteurs de la pompe à protons, antagonistes du récepteur H2).
Agents dont les concentrations plasmatiques peuvent être modifiées par le zanubrutinib
Le zanubrutinib est un inducteur léger du CYP3A et du CYP2C19. L'utilisation concomitante du zanubrutinib peut réduire les concentrations plasmatiques de ces médicaments substrats.
Substrats du CYP3A
La co-administration de plusieurs doses de zanubrutinib a diminué la Cmax du midazolam (substrat du CYP3A) de 30 % et son ASC de 47 %. Il convient d'utiliser avec prudence les médicaments à index thérapeutique étroit, qui sont métabolisés par le CYP3A (par exemple, alfentanil, ciclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, et tacrolimus) car le zanubrutinib peut réduire les expositions plasmatiques de ces médicaments.
Substrats du CYP2C19
La co-administration de plusieurs doses de zanubrutinib a diminué la Cmax de l'oméprazole (substrat du CYP2C19) de 20 % et son ASC de 36 %. Il convient d'utiliser avec prudence les médicaments à index thérapeutique étroit, qui sont métabolisés par le CYP2C19 (par exemple, S-méphénytoïne) car le zanubrutinib peut réduire les expositions plasmatiques de ces médicaments.
Autre substrats de CYP
Aucune différence cliniquement significative n'a été observée sur la pharmacocinétique de la S- warfarine (substrat du CYP2C9) co-administrées avec le zanubrutinib.
Co-administration avec des substrats/inhibiteurs de transporteur
La co-administration de plusieurs doses de zanubrutinib a augmenté la Cmax de la digoxine (substrat de la
P-gp) de 34 % et son ASC de 11 %. Aucune différence cliniquement
significative n'a été observée sur la pharmacocinétique de la
rosuvastatine (substrat de la BCRP) co-administrée avec le zanubrutinib.
La co-administration de substrats oraux de la P-gp ayant un index
thérapeutique étroit (par exemple, digoxine) doit être réalisée avec
prudence car le zanubrutinib peut augmenter leur concentration.
Le traitement par ce médicament doit être instauré et supervisé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Posologie
La dose quotidienne totale recommandée de zanubrutinib est de 320 mg. La dose quotidienne peut être prise une fois par jour (quatre gélules de 80 mg) ou divisée en deux prises de 160 mg deux fois par jour (deux gélules de 80 mg). Le traitement par Brukinsa doit être poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
BRUKINSA en association avec l'obinutuzumab
Le zanubrutinib doit être administré par voie orale avant la perfusion d'obinutuzumab. La dose recommandée est de 1 000 mg d'obinutuzumab par voie intraveineuse les jours 1, 8 et 15 du cycle 1, et le jour 1 de chaque cycle de 28 jours des cycles 2 à 6. À la discrétion du médecin, l'obinutuzumab peut être administré à raison de 100 mg le jour 1 et de 900 mg le jour 2 du cycle 1 au lieu de 1 000 mg le jour 1 du cycle 1. Un traitement d'entretien par obinutuzumab (une perfusion tous les deux mois pendant un maximum de deux ans) peut être prescrit. Se référer au RCP de l'obinutuzumab pour des informations supplémentaires sur la posologie, y compris la prémédication avant chaque perfusion.
Modifications de la dose en raison d'effets indésirables
Les modifications recommandées de la dose de zanubrutinib en raison d'effets indésirables de grade 3 ou plus sont indiquées dans le tableau 1.
Tableau 1 : modifications posologiques recommandées en raison d'effets indésirables
|
Effet indésirable |
Apparition de l'effet indésirable |
Modification de la dose (dose initiale : 320 mg une fois par jour ou 160 mg deux fois par jour) |
|
Toxicités non hématologiques de grade ≥ 3 Neutropénie fébrile de grade 3
Thrombopénie de grade 3 accompagnée d'un saignement important
Neutropénie de grade 4 (durant > 10 jours consécutifs)
Thrombopénie de grade 4 (durant > 10 jours consécutifs) |
Première |
Interrompre BRUKINSA Une fois la toxicité réduite à un grade ≤ 1 ou au niveau de référence : reprendre le traitement à 320 mg une fois par jour ou 160 mg deux fois par jour |
|
Deuxième |
Interrompre BRUKINSA Une fois la toxicité réduite à un grade ≤ 1 ou au niveau de référence : reprendre le traitement à 160 mg une fois par jour ou 80 mg deux fois par jour |
|
|
Troisième |
Interrompre BRUKINSA Une fois la toxicité réduite à un grade ≤ 1 ou au niveau de référence : reprendre le traitement à 80 mg une fois par jour |
|
|
Quatrième |
Arrêter définitivement BRUKINSA |
Une lymphocytose asymptomatique ne doit pas être considérée comme un effet indésirable, et les patients concernés doivent continuer à prendre BRUKINSA.
Pour la modification de la dose d'obinutuzumab en cas d'effets indésirables, se référer au RCP de l'obinutuzumab.
Modifications de la dose en cas de traitement concomitant
Modifications de la dose en cas d'utilisation d'inhibiteurs ou d'inducteurs de CYP3A (voir rubriques Mises en garde spéciales et précautions d'emploi, Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques) :
Tableau 2 : modifications de la dose recommandée en cas de co-administration avec d'autres médicaments
|
CYP3A |
Médicament co-administré |
Dose recommandée |
|
Inhibition |
Inhibiteur puissant du CYP3A (par exemple, posaconazole, voriconazole, kétoconazole, itraconazole, clarithromycine, indinavir, lopinavir, ritonavir, télaprévir)
Inhibiteur modéré du CYP3A (par exemple, érythromycine, ciprofloxacine, diltiazem, dronédarone, fluconazole, vérapamil, aprépitant, imatinib, jus de pamplemousse, oranges amères) |
80 mg une fois par jour
80 mg une fois par jour |
|
Induction |
Inducteur puissant du CYP3A (par exemple, carbamazépine, phénytoïne, rifampine, millepertuis) Inducteur modéré du CYP3A (par exemple, bosentan, éfavirenz, étravirine, modafinil, nafcilline) |
Éviter l'utilisation concomitante ; envisager d'autres traitements présentant une induction moindre du CYP3A |
Dose manquée
Il conviendra de ne pas prendre de dose double pour compenser une dose oubliée. Si une dose n'est pas prise au moment prévu, il faudra prendre la dose suivante selon le calendrier normal.
Populations particulières
Personnes âgées
Aucun ajustement posologique spécifique n'est nécessaire chez les patients âgés (≥ 65 ans).
Insuffisance rénale
Aucune modification de la dose n'est recommandée chez les patients présentant une insuffisance rénale légère à modérée (clairance de la créatinine ≥ 30 ml/min, estimée par l'équation de CockcroftGault). On dispose de données limitées chez les patients présentant une insuffisance rénale sévère et une insuffisance rénale chronique terminale (IRCT) (n = 12). Les patients avec une atteinte sévère de la fonction rénale (clairance de la créatinine < 30 ml/min) ou sous dialyse doivent être mis sous surveillance quant aux effets indésirables (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune modification de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique légère (classe Child Pugh A) ou modérée (classe Child Pugh B). Des patients présentant une insuffisance hépatique légère ou modérée ont été traités dans les études cliniques portant sur BRUKINSA. La dose recommandée de BRUKINSA pour les patients présentant une insuffisance hépatique sévère (classe Child Pugh C) est de 80 mg par voie orale deux fois par jour. La sécurité d'emploi de BRUKINSA n'a pas été évaluée chez les patients atteints d'insuffisance hépatique sévère. Surveiller étroitement les effets indésirables de BRUKINSA chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de BRUKINSA chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
BRUKINSA est administré par voie orale. Les gélules peuvent être prises avec ou sans nourriture. Il convient d'informer les patients d'avaler les gélules entières avec de l'eau, de ne pas ouvrir, rompre ni mâcher les gélules.
Durée de conservation :
3 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Il n'existe pas d'antidote spécifique de BRUKINSA. Les patients présentant un surdosage doivent être étroitement surveillés et un traitement symptomatique approprié doit être mis en place.
Classe pharmacothérapeutique : Agents antinéoplasiques, inhibiteurs de la tyrosine kinase de Bruton, Code ATC : L01EL03.
Mécanisme d'action
Le zanubrutinib est un inhibiteur de la tyrosine kinase de Bruton (BTK). Le zanutrutinib forme une liaison covalente avec un résidu cystéine sur le site actif de la BTK, ce qui entraîne l'inhibition de son activité. La BTK est une molécule de signalisation du récepteur d'antigène des lymphocytes B (BCR) et des voies des récepteurs de cytokines. Dans les lymphocytes B, la signalisation de la BTK entraîne l'activation des voies nécessaires à la prolifération, la circulation, la chimiotaxie et l'adhérence des lymphocytes B.
Effets pharmacodynamiques
Occupation de la BTK dans les cellules mononucléées
du sang périphérique et biopsies de ganglions lymphatiques
Le
taux d'occupation médian de la BTK à l'état d'équilibre dans les cellules mononucléées du sang périphérique s'est maintenu à 100 %
sur 24 heures à une dose quotidienne totale de 320 mg chez les patients
atteints de tumeurs malignes impliquant les lymphocytes B. Le taux d'occupation
médian de la BTK à l'état d'équilibre dans les ganglions lymphatiques variait
de 94 % à 100 % suite à l'administration de la posologie recommandée.
Effet sur l'intervalle QT/QTc et électrophysiologie cardiaque
Aux doses recommandées (320 mg une fois par jour ou 160 mg deux fois par jour), aucun effet pertinent sur le plan clinique n'a été observé sur l'intervalle QTc. À une dose unique correspondant à 1,5 fois la dose maximale recommandée (480 mg), le zanubrutinib n'a pas allongé l'intervalle QT de façon pertinente sur le plan clinique (c.-à-d., ≥ 10 ms).
Efficacité et sécurité cliniques
Patients atteints de macroglobulinémie de Waldenström (MW)
La
sécurité d'emploi et l'efficacité de BRUKINSA dans la MW ont été évaluées dans
une étude randomisée, en ouvert, multicentrique comparant le zanubrutinib et l'ibrutinib
(étude ASPEN, BGB- 3111-302) chez des patients qui étaient naïfs d'inhibiteurs
de la BTK. Les patients éligibles étaient âgés d'au moins 18 ans et
présentaient un diagnostic clinique et histologique certain de MW en
rechute/réfractaire ou naïfs de traitement et considérés inéligibles à une
chimio-immunothérapie standard selon leur médecin traitant. Les patients
devaient satisfaire à au moins un critère de traitement selon les critères du
groupe de consensus du Septième atelier international sur la macroglobulinémie
de Waldenström (IWWM) et présenter une maladie
mesurable, définie par un taux sérique d'IgM > 0,5
g/dl. Les patients porteurs de la mutation MYD88 (MYD88 MUT) ont été affectés à
la cohorte 1 (N = 201) et ont été randomisés selon un rapport de 1:1 pour
recevoir le zanubrutinib 160 mg deux fois par jour
(bras A) ou l'ibrutinib 420 mg une fois par jour
(bras B) jusqu'à la progression de la maladie ou l'apparition d'une toxicité
inacceptable. Les patients porteurs du type sauvage de MYD88 (MYD88WT)
déterminé par séquençage génétique (dont la présence est estimée à environ 10 %
des patients inclus), ont été inclus dans la cohorte 2 (N = 28) et ont reçu le zanubrutinib 160 mg deux fois par jour dans un troisième
bras de l'étude non randomisé (bras C).
Dans la cohorte 1 (MYD88MUT), l'âge médian était de 70 ans (étendue, 38 à 90 ans), 71 % et 60 % des patients traités par ibrutinib et zanubrutinib, respectivement, étaient âgés de > 65 ans, 33 % des patients du bras zanubrutinib et 22 % du bras ibrutinib étaient âgés de > 75 ans, 67 % étaient des hommes et 91 % étaient de type européen. À l'entrée dans l'étude, la valeur obtenue par les patients dans le système de score pronostique international (score IPSS), était élevée pour 44 % des patients dans le bras ibrutinib et 46 % des patients dans le bras zanubrutinib. Cent soixante-quatre patients présentaient une maladie en rechute ou réfractaire ; le nombre médian de traitements antérieurs était de 1 (étendue, 1 à 8).
La principale mesure de résultat était le taux de réponse complète (RC) ou de très bonne réponse partielle (TBRP), évalué par un comité d'examen indépendant (CEI) avec l'adaptation des critères de réponse mis à jour lors du sixième IWWM. Les critères secondaires d'évaluation pour la cohorte 1 comprenaient le taux de réponse majeure (TRM), la durée de réponse, le taux de RC ou de TBRP déterminé par l'investigateur, et la survie sans progression (SSP).
L'évaluation de la supériorité du critère d'évaluation principal du taux de TBRP ou de RC a nécessité l'évaluation du groupe d'analyse en rechute/réfractaire avant l'évaluation du groupe d'analyse ITT. La durée médiane du suivi était de 19,4 mois.
Chez les patients en rechute/réfractaires, 19,8 % et 28,9 % ont atteint une TBRP ou une RC dans les bras ibrutinib et zanubrutinib, respectivement. Le critère d'évaluation principal n'était pas significatif dans le groupe d'analyse en rechute/réfractaire (p bilatérale = 0,1160). Le tableau 5 résume les réponses évaluées par l'IRC pour les groupes d'analyse en rechute/réfractaire et ITT. Pour le zanubritinib, les réponses ont été observées au sein des sous-groupes, notamment les patients MYD88WT (Cohorte 2), dont la TBRP ou RC était de 26,9 % et le TRM de 50 %.
Tableau 5 : analyse de la réponse de la maladie selon un comité d'examen indépendant (étude ASPEN)
| Catégorie de réponse | En rechute/réfractaire | ITT | ||
|
Ibrutinib N = 81 |
Zanubrutinib N = 83 |
Ibrutinib N = 99 |
Zanubrutinib N = 102 |
|
|
Durée médiane du suivi,
mois (plage) |
18,79 (0,5 ; 30,0) |
18,73 (0,4 ; 28,7) |
19,38 (0,5 ; 31,1) |
19,47 (0,4 ; 31,2) |
| RC | 0 (0,0) | 0 (0,0) | 0 (0,0) | 0 (0,0) |
| TBRP | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
| RP | 49 (60,5) | 41 (49,4) | 58 (58,6) | 50 (49,0) |
| Taux de TBRP ou RC, n (%) | 16 (19,8) | 24 (28,9) | 19 (19,2) | 29 (28,4) |
| IC à 95 %a | (11,7 ; 30,1) | (19,5 ; 39,9) | (12,0 ; 28,3) | (19,9 ; 38,2) |
| Différence de risque (%)b | 10,7 | 10,2 | ||
| IC à 95 %a | (-2,5, 23,9) | (-1,5, 22,0) | ||
| valeur de pc | 0,1160 | |||
| TRM (RP ou mieux), n (%) | 65 (80,2) | 65 (78,3) | 77 (77,8) | 79 (77,5) |
| IC à 95 %a | (69,9 ; 88,3) | (67,9 ; 86,6) | (68,3 ; 85,5) | (68,1 ; 85,1) |
| Différence de risque (%)b | -3,5 | -0,5 | ||
| IC à 95 % | (-16,0 ; 9,0) | (-12,2 ; 11,1) | ||
| Durée de la réponse majeure | ||||
|
Taux sans événement à 18 mois, % (IC à 95 %a )d |
85,6 (73,1 ; 92,6) |
87,0 (72,5 ; 94,1) |
87,9 (77,0 ; 93,8) |
85,2 (71,7 ; 92,6) |
Les
pourcentages sont basés sur N.
a Intervalle de confiance bilatéral à 95 % de Clopper-Pearson.
b Différence de risque fréquent de Mantel-Haenszel avec l'intervalle de confiance à 95 % calculé en utilisant une approximation normale et erreur type de Sato stratifiée selon les facteurs de stratification de l'IRT (les strates CXCR4 de type sauvage et inconnu sont combinées) et la tranche d'âge (≤ 65 et > 65). L'ibrutinib est le bras de référence.
c Basé sur un test CMH stratifié selon les facteurs de stratification de l'IRT (les strates CXCR4 de type sauvage et inconnu sont combinées) et la tranche d'âge (≤ 65 et > 65).
d Les taux sans événement sont estimés par la méthode de Kaplan-Meier avec des IC à 95 % estimés à l'aide de la formule de Greenwood.
a Intervalle de confiance bilatéral à 95 % de Clopper-Pearson.
b Différence de risque fréquent de Mantel-Haenszel avec l'intervalle de confiance à 95 % calculé en utilisant une approximation normale et erreur type de Sato stratifiée selon les facteurs de stratification de l'IRT (les strates CXCR4 de type sauvage et inconnu sont combinées) et la tranche d'âge (≤ 65 et > 65). L'ibrutinib est le bras de référence.
c Basé sur un test CMH stratifié selon les facteurs de stratification de l'IRT (les strates CXCR4 de type sauvage et inconnu sont combinées) et la tranche d'âge (≤ 65 et > 65).
d Les taux sans événement sont estimés par la méthode de Kaplan-Meier avec des IC à 95 % estimés à l'aide de la formule de Greenwood.
D'après une mise à jour de la date de clôture des données, le taux sans événement de la survie sans progression, d'après l'évaluation de l'investigateur, était de 77,6 % versus 84,9 % à 30 mois (ibrutinib versus zanubrutinib), avec un risque relatif global estimé de 0,734 (IC à 95 % : 0,380 ; 1,415).
Patients atteints de lymphome de la zone marginale (LZM)
L'efficacité du zanubrutinib a été évaluée dans le cadre d'un essai de phase 2 en ouvert, multicentrique et à bras unique, mené auprès de 68 patients atteints de LZM ayant reçu au moins un traitement antérieur à base d'anticorps anti-CD20 (Étude MAGNOLIA, BGB-3111-214). Vingt-six (38,2 %) patients présentaient un LZM extra-ganglionnaire, 26 (38,2 %) présentaient un LZM ganglionnaire, 12 (17,6 %) présentaient un LZM splénique et le sous-type n'était pas connu chez 4 patients (6 %). Le zanubrutinib a été administré par voie orale à raison d'une dose de 160 mg deux fois par jour jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable. L'âge médian des patients était de 70 ans (tranche d'âge : 37 à 95) et 53 % étaient des hommes. Le délai médian depuis le diagnostic initial était de 61,5 mois (plage : 2,0 à 353,6). Le nombre médian de traitements antérieurs était de 2 (plage : 1 à 6), dont 27,9 % de patients ayant eu au moins 3 lignes de traitement systémique ; 98,5 % (n = 67) des patients avaient reçu une chimiothérapie antérieure à base de rituximab et 85,3 % (n = 58) des patients avaient reçu un traitement antérieur à base d'agents alkylants ; 5,9 % (n = 4) avaient reçu une greffe de cellules souches antérieure. Soixante-trois (92,6 %) des patients avaient un indice de performance ECOG à la référence de 0 ou 1. Vingt-deux (32,4 %) patients présentaient une maladie réfractaire à l'entrée dans l'étude.
La réponse tumorale était évaluée en fonction de la Classification de Lugano 2014, et le critère d'évaluation principal de l'efficacité était le taux de réponse globale, évalué par un comité d'examen indépendant (CEI) (Tableau 6).
Tableau 6 : Résultats d'efficacité chez les patients atteints de LZM selon le comité d'évaluation indépendant (étude MAGNOLIA)
|
Étude BGB-3111-214 (N = 66)* |
|
| TRG (95 % IC) | 68 % (55,6 - 79,1) |
| RC | 26 % |
| RP | 42 % |
| DR médiane en mois (IC à 95 %) | NE (25,0, NE) |
| Taux sans événementb pour la DR à 24 mois, % (IC à 95 %) | 72,9 (54,4, 84,9) |
| Suivi médian dans l'étude en mois (min, max) | 28,04 (1,64, 32,89) |
a Deux patients de l'étude BGB-3111-214 n'étaient pas
évaluables pour l'efficacité en raison de la confirmation par le laboratoire
central de la transformation du LZM en lymphome diffus à grandes cellules B.
b Les taux sans événement ont été estimés par la méthode Kaplan-Meier avec des IC à 95 % estimés à l'aide de la formule de Greenwood.
TRG : taux de réponse globale ; RC : réponse complète ; RP : réponse partielle ; DR : durée de la réponse ; IC : indice de confiance ; NE : non estimable
b Les taux sans événement ont été estimés par la méthode Kaplan-Meier avec des IC à 95 % estimés à l'aide de la formule de Greenwood.
TRG : taux de réponse globale ; RC : réponse complète ; RP : réponse partielle ; DR : durée de la réponse ; IC : indice de confiance ; NE : non estimable
Dans
l'étude BGB-3111-214, le délai médian jusqu'à la réponse était de 2,79 mois
(plage : 1,7 à 11,1 mois). Après une durée de suivi médiane dans l'étude de
28,04 mois (plage : 1,64 à 32,89 mois) la durée de la réponse (DR) médiane
évaluée par le CEI n'a pas été atteinte (IC à 95 % : 25,0 mois à NE) et un
total de 72,9 % (IC à 95 % : 54,4 à 84,9) des répondeurs a été estimé sans
événement à24 mois après la réponse initiale.
Les taux de réponse globale observés étaient similaires entre les trois
différents sous-types de LZM (extra-ganglionnaire, ganglionnaire et splénique).
Patients atteints de leucémie lymphoïde chronique (LLC)
L'efficacité de BRUKINSA chez des patients atteints de LLC a été évaluée dans deux essais randomisés contrôlés.
Étude SEQUOIA (BGB-3111-304) : étude internationale de phase III, en ouvert, randomisée, portant sur le zanubrutinib comparé à la bendamustine plus rituximab (BR) chez des patients atteints de LLC naïfs de traitement.
L'étude SEQUOIA (BGB-3111-304) est un essai de phase III multicentrique, randomisé, en ouvert, avec contrôle actif portant sur le zanubrutinib en monothérapie et la bendamustine en association avec le rituximab chez 479 patients atteints de LLC naïfs de traitement, sans délétion 17p (del(17p)) (bras A et B ; Cohorte 1). Le Bras C (Cohorte 2) est un essai multicentrique à bras unique portant sur une monothérapie de zanubrutinib chez 110 patients atteints de LLC naïfs de traitement et présentant une del(17p) confirmée centralement.
Les deux cohortes ont recruté des patients âgés de 65 ans ou plus ainsi que des patients âgés de 18 à 65 ans, n'étant pas en mesure de recevoir une chimio-immunothérapie par fludarabine, cyclophosphamide et rituximab (FCR).
Les caractéristiques démographiques et de référence étaient globalement équilibrées entre le bras A (zanubrutinib) et le bras B (BR) de la Cohorte 1. Dans les deux bras, l'âge médian était de 70,0 ans, avec une proportion légèrement supérieure de patients âgés de ≥ 75 ans (26,1 %) dans le bras A comparativement au bras B (22,3 %) et une proportion légèrement inférieure de patients âgés de 65-75 ans (55,2 %) dans le bras A comparativement au bras B (58,4 %). Dans la Cohorte 1, 92,7 % des patients avaient un indice de performance ECOG à la référence de 0 ou 1 (93,7 % dans le bras A et 91,6 % dans le bras B). Dans la Cohorte 2 (bras C zanubrutinib), 87,3 % des patients avaient un indice de performance ECOG à la référence de 0 ou 1.
Les caractéristiques démographiques et de référence étaient également généralement similaires entre le bras A (zanubrutinib) dans la Cohorte 1 et le bras C (zanubrutinib) dans la Cohorte 2.
Dans la Cohorte 1, la randomisation était stratifiée selon l'âge (< 65 ans vs ≥ 65 ans), le stade de Binet (C versus A ou B), le statut mutationnel (muté vs non muté) des régions variables des chaînes lourdes des immunoglobulines (immunoglobulin variable region heavy chain, IGHV) et la région géographique (Amérique du Nord versus Europe versus Asie Pacifique). Un total de 479 patients a été randomisé (population d'analyse en intention de traiter [ITT), 241 pour recevoir le zanubrutinib en monothérapie continue et 238 pour recevoir 6 cycles de traitement par bendamustine et rituximab (BR).
Dans la Cohorte 1, les patients dans le bras A zanubrutinib ont reçu 160 mg deux fois par jour jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable. Dans le bras B, les patients ont reçu la bendamustine à une dose de 90 mg/m2/jour les 2 premiers jours de chaque cycle pendant 6 cycles et le rituximab à une dose de 375 mg/m2 pendant le Cycle 1, et à une dose de 500 mg/m2 pendant les Cycles 2 à 6. Chaque cycle de traitement comprenait environ 28 jours. Dans la Cohorte 2 (bras C), les patients ont reçu le zanubrutinib 160 mg deux fois par jour jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Pour la Cohorte 1, le critère d'évaluation principal était la survie sans progression (SSP), évaluée par un comité d'évaluation central indépendant (CEI). Les critères d'évaluation secondaires comprenaient le taux de réponse global (TRG), basé sur l'évaluation du CEI.
Dans la Cohorte 1, la durée médiane du suivi pour la SSP était de 25,0 mois (intervalle : 0,0 à 41,4). Le taux de SSP à 24 mois était de 85,5 % (IC à 95 % : 80,1, 89,6) pour le zanubrutinib et de 69,5 % (IC à 95 % : 62,4, 75,5) pour BR. Dans la Cohorte 2, la durée médiane du suivi pour la SSP était de 27,9 mois (intervalle : 1,0 à 38,8) et le taux de SSP à 24 mois était de 88,9 % (IC à 95 % : 81,3, 93,6). Le TRG évalué par le CEI dans la Cohorte 2 était de 90,0 % (IC à 95 % : 82,8, 94,9). Le délai médian jusqu'à une réponse partielle ou supérieure évaluée par le CEI était de 2,89 mois (intervalle : 1,8, 14,2) et de 2,86 mois (intervalle : 1,9, 13,9) dans le bras zanubrutinib de la Cohorte 1 et de la Cohorte 2, respectivement.
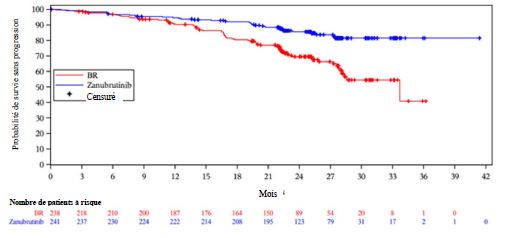
Les résultats d'efficacité pour la Cohorte 1 sont présentés dans le Tableau 7. Les courbes de Kaplan- Meier pour la SSP pour les deux bras de la Cohorte 1 sont présentées dans la Figure 1.
Tableau 7 : Résultats d'efficacité dans l'étude SEQUOIA
|
Cohorte 1*
Patients sans Del(17p) |
||
| Critère d'évaluation |
Zanubrutinib (N = 241) |
Bendamustine +
Rituximab (N = 238) |
| Survie sans progression† | ||
| Nombre d'événements, n (%) | 36 (14,9) | 71 (29,8) |
| Progression de la maladie, n (%) | 27 (11,2) | 59 (24,8) |
| Décès, n (%) | 9 (3,7) | 12 (5,0) |
| Médiane (IC à 95 %), moisa | NE (NE, NE) | 33,7 (28,1, NE) |
| Ratio de risque (IC à 95 %)b | 0,42 (0,28, 0,63) | |
| Valeur pc | < 0,0001 | |
|
Taux
de réponse global† % (95% CI) |
94,6 %
(91,0, 97,1) |
85,3 %
(80,1, 89,5) |
Taux
de réponse globale : RC+RCi+RPn+RP+RP-L, RC : réponse
complète, RCi : réponse complète avec récupération
hématopoïétique incomplète, RPn : réponse partielle
nodulaire, RP : réponse partielle, RP-L : réponse partielle avec lymphocytome, IC : intervalle de confiance, NE : non
estimable, la durée médiane du suivi pour la SSP était de 25,0 mois (IC à 95 %
: 24,6, 25,2).
* Population d'analyse en ITT
† Évalué par un comité d'évaluation central indépendant.
a Basé sur une estimation Kaplan-Meier.
b Basé sur un modèle de régression de Cox stratifié avec bendamustine + rituximab comme groupe de référence.
c Basé sur un test log-rank stratifié.
* Population d'analyse en ITT
† Évalué par un comité d'évaluation central indépendant.
a Basé sur une estimation Kaplan-Meier.
b Basé sur un modèle de régression de Cox stratifié avec bendamustine + rituximab comme groupe de référence.
c Basé sur un test log-rank stratifié.
Lors d'une analyse ad hoc mise à jour avec un suivi médian de 33,5 mois pour la SSP, l'investigateur a déterminé que la SSP reste cohérente avec celle de l'analyse primaire avec un RR de 0,33 (IC à 95 % : 0,22 à 0,48, p descriptif < 0,0001) dans le bras zanubrutinib par rapport au bras BR. La SSP médiane n'a pas été atteinte dans le bras zanubrutinib et était de 39,2 mois dans le bras BR. À 36 mois après la randomisation, 83,6 % des patients traités par zanubrutinib et 55,1 % des patients traités par BR ont été estimés comme étant sans progression et en vie. Avec un suivi médian de 35,8 mois, la SG médiane n'a été atteinte dans aucun des deux bras, l'estimation du taux de SG à 36 mois était de 90,9 % (IC à 95 % : 86,3 à 94,0) dans le bras zanubrutinib et de 89,5 % (IC à 95 % : 84,2 à 93,1) dans le bras BR, respectivement.
Figure 1 : Courbe de Kaplan-Meier de la SSP évaluée par le CEI dans la Cohorte 1 de l'étude SEQUOIA (population en ITT)
Étude ALPINE (BGB-3111-305) : étude de phase III,
randomisée portant sur le zanubrutinib comparé à l'ibrutinib chez des patients atteints de LLC en rechute/réfractaire
(R/R)
L'étude ALPINE (BGB-3111-305) est un essai de Phase III, randomisé, multicentrique, en ouvert avec contrôle actif. Celui-ci a recruté 652 patients atteints de LLC en rechute ou réfractaire après au moins un traitement systémique antérieur. Les patients ont été randomisés afin de recevoir soit le zanubrutinib 160 mg par voie orale deux fois par jour, soit l'ibrutinib 420 mg par voie orale une fois par jour, poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
La randomisation était stratifiée selon l'âge (< 65 ans versus ≥ 65 ans), la région géographique (Chine versus hors Chine), le statut réfractaire (oui ou non) et le statut mutationnel de del(17p)/TP53 (présent ou absent).
Les données démographiques et les caractéristiques de la maladie à la référence étaient généralement équilibrées entre les différents bras de traitement de la population d'analyse en ITT ainsi que chez les 415 premiers patients randomisés.
Dans la population d'analyse en ITT, l'âge médian était de 67,0 ans dans le bras zanubrutinib et de 68,0 ans dans le bras ibrutinib. La majorité des patients dans les deux bras avait un indice de performance ECOG de 0 ou 1 (97,9 % dans le bras zanubrutinib ; 96,0 % dans le bras ibrutinib). Des données démographiques et caractéristiques de référence similaires ont été observées chez les 415 premiers patients randomisés. Le nombre médian de lignes de traitement systémique antérieures était de 1,0 dans le bras zanubrutinib (intervalle, 1 à 6) et de 1,0 dans le bras ibrutinib (intervalle, 1 à 8) dans la population d'analyse en ITT ainsi que chez les 415 premiers patients randomisés.
Les patients précédemment traités par un inhibiteur de BTK ont été exclus de l'étude 305 et des données limitées sont disponibles pour le zanubrutinib après un traitement antérieur par inhibiteur de BCL-2.
Sur un total de 652 patients, 327 ont été affectés au zanubrutinib en monothérapie, 325 à l'ibrutinib en monothérapie. L'évaluation de l'efficacité s'appuie sur l'analyse intermédiaire pré-spécifiée des 415 premiers patients randomisés de la population en ITT. Sur ceux-ci, 207 ont été randomisés pour recevoir le zanubrutinib en monothérapie, et 208 l'ibrutinib en monothérapie. Les résultats d'efficacité sont présentés dans le Tableau 8.
Le critère d'évaluation principal était le taux de réponse globale (TRG, défini comme une réponse partielle ou meilleure).
À l'analyse intermédiaire préétablie du TRG chez les 415 premiers patients randomisés, le zanubrutinib a démontré une non-infériorité (valeur unilatérale de p < 0,0001) et une supériorité (valeur bilatérale de p = 0,0006) par rapport à l'ibrutinib en ce qui concerne le TRG, critère d'évaluation principal spécifié dans le protocole, évalué par l'investigateur. La réponse telle que déterminée par le CEI a également démontré une non-infériorité du zanubrutinib par rapport à l'ibrutinib (valeur unilatérale de p < 0,0001). À l'analyse finale du TRG, le TRG évalué par l'investigateur continue d'être supérieur (79,5 % versus 71,1 %) dans le bras zanubrutinib comparé au bras ibrutinib (valeur descriptive de p = 0,0133) ; le TRG déterminé par le CEI était également significativement supérieur dans le bras zanubrutinib comparativement au bras ibrutinib, démontrant une supériorité (80,4 % versus 72,9 %, respectivement ; valeur bilatérale de p = 0,0264).
Tableau 8 : Résultats d'efficacité dans l'étude ALPINE (Analyse intermédiaire pré-spécifiée des 415 premiers patients randomisés) selon l'évaluation de l'investigateur (critère d'évaluation principal défini dans le protocole) et du CEI
|
Évalué par l'investigateur
(critère d'évaluation principal défini dans le protocole) |
Évalué par le CEI | |||
| Critère d'évaluation |
Zanubrutinib (N = 207) |
Ibrutinib (N = 208) |
Zanubrutinib
(N = 207) |
Ibrutinib (N = 208) |
|
Taux
de réponse globale§
n (%) (IC à 95 %) |
162 (78,3) (72,0, 83,7) |
130 (62,5) (55,5, 69,1) |
153 (76,3) (69,9, 81,9) |
134 (64,4) (57,5, 70,9) |
| Taux de réponse a (IC à 95 %) | 1,25 (1,10, 1,41) | 1,17 (1,04, 1,33) | ||
| Non-inférioritéb | Valeur unilatérale de p < 0,0001 | Valeur unilatérale de p < 0,0001 | ||
| Supérioritéc | Valeur bilatérale de p 0,0006 | Valeur bilatérale de p 0,0121 | ||
|
Durée
de la réponsed : taux sans événement à 12 mois, % (IC à 95 %) |
89,8 (78,1, 95,4) |
77,9 (64,7, 86,7) |
90,3 (82,3, 94,8) |
78,0 (66,1, 86,2) |
Taux
de réponse globale : RC + RCi + RPn
+ RP, RC : réponse complète, RCi: réponse complète
avec récupération hématopoïétique incomplète, RPn :
réponse partielle nodulaire, RP : réponse partielle, IC : intervalle de
confiance
La durée médiane de la réponse telle qu'évaluée par l'investigateur n'a pas été atteinte dans le bras zanubrutinib lors de l'analyse intermédiaire, la durée médiane du suivi dans l'étude était de 15,31 mois (intervalle : 0,1, 23,1) dans le bras zanubrutinib et de 15,43 mois (intervalle : 0,1, 26,0) dans le bras ibrutinib.
§ L'évaluation de l'hypothèse de non-infériorité du TRG lors de l'analyse intermédiaire est basée sur les 415 premiers patients randomisés uniquement avec un niveau de signification unilatéral de 0,005.
a Ratio de réponse : ratio estimé du taux de réponse globale dans le bras zanubrutinib divisé par celui du bras ibrutinib.
b Test stratifié selon un taux de réponse nul de 0,8558.
c Test de Cochran-Mantel-Haenszel stratifié.
d Estimation Kaplan-Meier.
La durée médiane de la réponse telle qu'évaluée par l'investigateur n'a pas été atteinte dans le bras zanubrutinib lors de l'analyse intermédiaire, la durée médiane du suivi dans l'étude était de 15,31 mois (intervalle : 0,1, 23,1) dans le bras zanubrutinib et de 15,43 mois (intervalle : 0,1, 26,0) dans le bras ibrutinib.
§ L'évaluation de l'hypothèse de non-infériorité du TRG lors de l'analyse intermédiaire est basée sur les 415 premiers patients randomisés uniquement avec un niveau de signification unilatéral de 0,005.
a Ratio de réponse : ratio estimé du taux de réponse globale dans le bras zanubrutinib divisé par celui du bras ibrutinib.
b Test stratifié selon un taux de réponse nul de 0,8558.
c Test de Cochran-Mantel-Haenszel stratifié.
d Estimation Kaplan-Meier.
Le délai médian jusqu'à la réponse, tel qu'évalué par l'investigateur lors de l'analyse intermédiaire du TRG chez les 415 premiers patients randomisés était de 5,59 mois (intervalle : 2,7, 14,1) dans le bras zanubrutinib et de 5,65 mois (intervalle : 2,8, 16,7) dans le bras ibrutinib. Les résultats évalués par le CEI étaient cohérents (5,55 mois vs. 5,63 mois dans les bras zanubrutinib et ibrutinib respectivement). Lors de l'analyse finale du TRG chez la totalité des 652 patients randomisés, le délai médian jusqu'à la réponse est demeuré inchangé (5,59 mois vs. 5,65 mois, selon l'évaluation de l'investigateur et 5,52 mois vs. 5,62 mois selon l'évaluation du CEI dans les bras zanubrutinib et ibrutinib respectivement).
Chez les patients porteurs de la mutation del(17p) parmi les 415 premiers patients randomisés, le TRG évalué par l'investigateur était de 83,3 % (IC à 95 % 62,5, 95,3 ; 20 patients sur 24) dans le bras zanubrutinib et de 53,8 % (IC à 95 % 33,4, 73,4 ; 14 patients sur 26) dans le bras ibrutinib. D'après l'évaluation du CEI, le TRG était de 79,2 % (IC à 95 % 57,8, 92,9 ; 19 patients sur 24) dans le bras zanubrutinib et de 61,5 % (IC à 95 % 40,6, 79,8 ; 16 patients sur 26) dans le bras ibrutinib. Lors de l'analyse finale du TRG chez la totalité des 652 patients randomisés, le TRG évalué par l'investigateur était de 86,7 % (IC à 95 % 73,2, 94,9 ; 39 patients sur 45 porteurs de la mutation del(17p)) dans le bras zanubrutinib et de 56,0 % (IC à 95 % 41,3, 70,0 ; 28 patients sur 50 porteurs de la mutation del(17p)) dans le bras ibrutinib. D'après l'évaluation du CEI, le TRG était de 86,7 % (IC à 95 % 73,2, 94,9 ; 39 patients sur 45 porteurs de la mutation del(17p)) dans le bras zanubrutinib et de 64,0 % (IC à 95 % 49,2, 77,1 ; 32 patients sur 50 porteurs de la mutation del(17p) dans le bras ibrutinib.
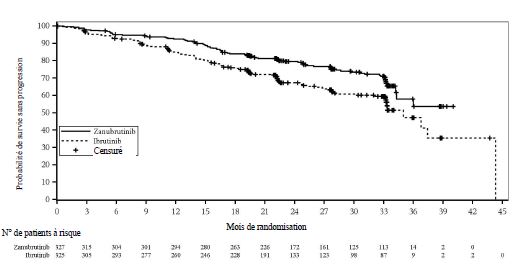
Au total, 652 patients ont été inclus au moment pré-spécifié de l'analyse finale de la SSP (date limite du 8 août 2022). La durée médiane de suivi de la SSP était de 28,1 mois selon l'évaluation de l'investigateur et de 30,7 mois selon l'évaluation du CEI. Le zanubrutinib a montré une supériorité concernant la SSP par rapport à l'ibrutinib selon l'évaluation de l'investigateur et du CEI. Les résultats d'efficacité concernant la SSP sont présentés dans le tableau 9, et un diagramme de Kaplan-Meier selon l'évaluation du CEI est fourni dans la figure 2.
Tableau 9 : Résultats d'efficacité dans l'étude ALPINE (analyse finale pré-spécifiée de la SSP de l'ensemble des 652 patients randomisés) selon l'évaluation de l'investigateur et du CEI (date limite du 8 août 2022)
| Critère d'évaluation | Évaluation par l'investigateur | Évaluation indépendante* | ||
|
Zanubrutinib
(N = 327) |
Ibrutinib
(N = 325) |
Zanubrutinib (N = 327) |
Ibrutinib (N = 325) |
|
| Survie sans progression | ||||
| Événements, n (%) | 87 (26,6) | 118 (36,3) | 88 (26,9) | 120 (36,9) |
| Rapport de risquea (IC à95 %) | 0,65 (0,49 ; 0,86) | 0,65 (0,49 ; 0,86) | ||
| Valeur de p bilatéraleb | 0,0024 | 0,0024 | ||
a Basé sur un modèle de régression de Cox stratifié avec l'ibrutinib comme groupe de référence.
b Basé sur un test du log-rank stratifié.
b Basé sur un test du log-rank stratifié.
Figure 2 : Courbe de Kaplan-Meier de la survie sans progression par examen central indépendant (ITT) (date limite du 8 août 2022)
Chez les patients présentant une mutation del(17p)/TP53, le rapport de risque pour la survie sans progression selon l'évaluation de l'investigateur était de 0,53 (IC à 95 % : 0,31, 0,88). D'après l'examen indépendant, le rapport de risque était de 0,52 (IC à 95 % : 0,30, 0,88) (figure 3).
Figure 3 : Courbe de Kaplan-Meier de la survie sans progression selon l'examen central indépendant pour les patients présentant une mutation del(17p) ou TP53 (ITT) (date limite du 8 août 2022)
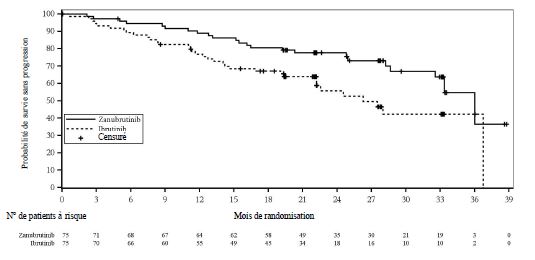
Avec un suivi médian estimé de 32,8 mois, la médiane de survie globale n'a été atteinte dans aucun des deux bras, 17 % des patients ayant présenté un événement.
Patients atteints de lymphome folliculaire (LF)
L'efficacité du zanubrutinib en association avec l'obinutuzumab par rapport à l'obinutuzumab a été évaluée dans l'étude ROSEWOOD (BGB-3111-212), une étude de phase 2 randomisée, en ouvert et multicentrique. Au total, 217 patients atteints d'un lymphome folliculaire (LF) de grade 1-3a en rechute (défini par une progression de la maladie après la fin du traitement le plus récent) ou réfractaire (défini par l'absence de RC ou de RP au traitement le plus récent) et ayant déjà reçu au moins deux traitements systémiques, dont un anticorps anti-CD20 et une polythérapie appropriée à base d'alkylants, ont été inclus dans l'étude. Les patients ont été randomisés selon un rapport 2:1 pour recevoir soit 160 mg de zanubrutinib par voie orale deux fois par jour jusqu'à la progression de la maladie ou une toxicité inacceptable, en association avec 1 000 mg d'obinutuzumab par voie intraveineuse (bras A), ou l'obinutuzumab en monothérapie (bras B). L'obinutuzumab a été administré les jours 1, 8 et 15 du premier cycle, puis le jour 1 des cycles 2 à 6. Chaque cycle a duré 28 jours. Les patients ont reçu en option une perfusion d'entretien d'obinutuzumab tous les deux cycles, pour un maximum de 20 doses.
Les patients randomisés dans le groupe obinutuzumab ont été autorisés à changer de groupe et à recevoir l'association zanubrutinib plus obinutuzumab en cas de maladie progressive ou d'absence de réponse (définie par une maladie stable comme meilleure réponse) après 12 cycles.
La randomisation a été stratifiée en fonction du nombre de lignes de traitement antérieures (2 à 3 versus ?3), du statut réfractaire au rituximab (oui versus non) et de la région géographique (Chine versus autres pays).
Les données démographiques à la référence et les caractéristiques de la maladie étaient généralement équilibrées entre le bras d'association de zanubrutinib et le bras de monothérapie d'obinutuzumab chez les 217 patients randomisés. L'âge médian était de 64 ans (de 31 à 88 ans), 49,8 % des patients étaient des hommes et 64,1 % étaient de race blanche. La plupart (97,2 %) des patients avaient un statut de performance ECOG de 0 ou 1.
Au moment de la sélection, la plupart des patients étaient au stade III ou IV d'Ann Arbor (179 patients [82,5 %]). Quatre-vingt-huit patients (40,6 %) présentaient une maladie volumineuse (définie comme > 1 lésion cible initiale mesurant > 5 cm de diamètre). Cent vingt-trois patients (56,7 %) répondaient aux critères du GELF.
Le nombre médian de traitements anticancéreux antérieurs était de 3 lignes (intervalle : 2 à 11 lignes). La totalité des 217 patients a reçu plus de 2 lignes de traitement antérieures comprenant du rituximab (en monothérapie ou en association avec une chimiothérapie), et 59 des 217 patients (27,2 %) ont reçu plus de 3 lignes de traitement antérieures. Sur les 217 patients, 114 (52,5 %) étaient réfractaires au rituximab (défini comme l'absence de réponse ou la progression au cours d'un traitement antérieur contenant du rituximab [en monothérapie ou en association avec une chimiothérapie], ou la progression dans les 6 mois suivant la dernière dose de rituximab, dans le cadre d'un traitement d'induction ou d'un traitement d'entretien). Douze patients (5,5 %) avaient déjà reçu de l'obinutuzumab.
Sur un total de 217 patients, 145 ont été randomisés dans le bras d'association de zanubrutinib et 72 dans le bras de monothérapie d'obinutuzumab. La durée médiane de suivi a été de 20,21 mois dans le bras de l'association zanubrutinib-obinutuzumab et de 20,40 mois dans le bras de monothérapie d'obinutuzumab. La durée médiane d'exposition au zanubrutinib était de 12,16 mois.
Sur les 72 patients randomisés dans le groupe obinutuzumab en monothérapie, 35 sont passés au traitement d'association.
Le critère principal d'efficacité était le taux de réponse globale (défini comme une réponse partielle ou une réponse complète) déterminé par un examen central indépendante en utilisant la classification de Lugano pour les LNH. Les principaux critères d'évaluation secondaires étaient la durée de la réponse (DOR), la survie sans progression (PFS) et la survie globale (OS).
Les résultats d'efficacité sont résumés dans le tableau 10 et la figure 4.
Tableau 10 : Résultats d'efficacité selon l'examen central indépendant (ITT) (étude ROSEWOOD)
|
Zanubrutinib +
Obinutuzumab (N = 145) n (%) |
Obinutuzumab (N = 72) n (%) |
|
|
Taux
de réponse global,
n (%) (IC à 95 %a) |
100 (69,0) (60,8, 76.,4) |
33 (45,8) (34,0 ; 58,0) |
| Valeur de Pb | 0,0012 | |
| CR | 57 (39,3) | 14 (19,4) |
| PR | 43 (29,7) | 19 (26,4) |
| Durée de la réponse (mois) | ||
| Médiane (IC à 95 %)c | NE (25,3 ; NE) | 14 (9,2 ; 25,1) |
| Taux de DR à 12 mois (IC à 95 %)d | 72,8 (62,1, 80,9) | 55,1 (34,4, 71,6) |
| Taux de DR à 18 mois (IC à 95 %)d | 69,3 (57,8 ; 78,2) | 41,9 (22,6 ; 60,1) |
| Survie sans progression (Mois) | ||
| Médiane (IC à 95 %)c | 28,0 (16,1, NE) | 10,4 (6,5 ; 13,8) |
Taux
de réponse global : CR + PR, CR: réponse complète, PR: réponse partielle
a Estimation selon la méthode Clopper-Pearson.
b Méthode Cochran-Mantel-Haenszel stratifiée en fonction du statut réfractaire au rituximab, du nombre de lignes de traitement antérieures et de la région géographique selon l'IRT.
c Médianes estimées par la méthode Kaplan-Meier ; IC à 95 % estimés par la méthode Brookmeyer et Crowley.
d Taux de DR estimés par la méthode de Kaplan-Meier ; IC à 95 % estimés à l'aide de la formule de Greenwood. La DR n'a pas été contrôlée contre l'erreur de type I et les IC sont nominaux par nature.
a Estimation selon la méthode Clopper-Pearson.
b Méthode Cochran-Mantel-Haenszel stratifiée en fonction du statut réfractaire au rituximab, du nombre de lignes de traitement antérieures et de la région géographique selon l'IRT.
c Médianes estimées par la méthode Kaplan-Meier ; IC à 95 % estimés par la méthode Brookmeyer et Crowley.
d Taux de DR estimés par la méthode de Kaplan-Meier ; IC à 95 % estimés à l'aide de la formule de Greenwood. La DR n'a pas été contrôlée contre l'erreur de type I et les IC sont nominaux par nature.
Figure 4 : Graphique de Kaplan-Meier de la survie sans progression selon l'examen central indépendant (ITT)
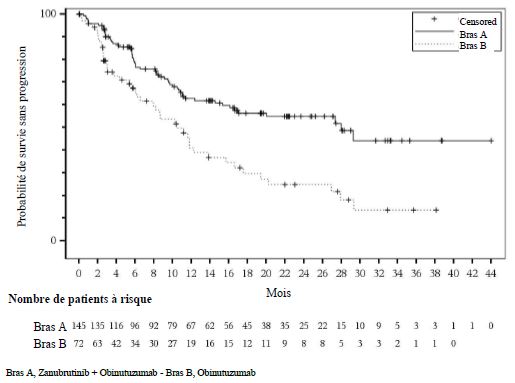
Survie
globale
Vingt-neuf
patients (20,0 %) du bras d'association et 22 patients (30,6 %) du bras de
monothérapie d'obinutuzumab sont décédés. À 18 mois,
les taux de survie globale étaient de 84,6 % (IC à 95 % : 77,1, 89,8) dans le
bras d'association et de 73,5 % (IC à 95 % : 60,7, 82,7) dans le bras de
monothérapie d'obinutuzumab. L'analyse de la SG peut
être faussée par le fait que 35 patients (48,6 %) sont passés du bras obinutuzumab en monothérapie au bras d'association.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec BRUKINSA dans tous les sous-groupes de la population pédiatrique pour le traitement du lymphome lymphoplasmocytaire et pour le traitement des néoplasmes à lymphocytes B matures (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La concentration plasmatique maximale en zanutrutinib (Cmax) et l'aire sous la courbe (ASC) de concentration plasmatique en médicament au fil du temps augmentent proportionnellement sur l'intervalle de dose de 40 mg à 320 mg (0,13 à 1 fois la dose quotidienne totale recommandée). Une accumulation systémique limitée de zanubrutinib a été observée après une administration répétée pendant une semaine.
La moyenne géométrique (% CV) de l'ASC quotidienne à l'état d'équilibre du zanutrutinib est de 2 099 (42 %) ng·h/ml après l'administration de 160 mg deux fois par jour et de 1 917 (59 %)ng·h/ml après l'administration de 320 mg une fois par jour. La moyenne géométrique (% CV) de la Cmax à l'état d'équilibre du zanutrutinib est de 299 (56 %) ng/ml après l'administration de 160 mg deux fois par jour et de 533 (55 %) ng/ml après l'administration de 320 mg une fois par jour.
Absorption
La tmax médiane du zanubrutinib est de 2 heures. Aucune différence cliniquement significative de l'ASC ou de la Cmax du zanutrutinib n'a été observée après l'administration d'un repas riche en matières grasses (environ 1 000 calories avec 50 % de la teneur totale en calories provenant des graisses) chez des sujets en bonne santé.
Distribution
La
moyenne géométrique (% CV) du volume de distribution apparente à l'état
d'équilibre du zanubrutinib pendant la phase terminale (Vz/F) était de
522 litres (71 %). La liaison du zanubrutinib aux protéines
plasmatiques représente environ 94 % et le rapport sang/plasma était
compris entre 0,7 et 0,8.
Métabolisme
Le zanutrutinib est principalement métabolisé par le cytochrome P450 (CYP) 3A.
Élimination
La demi-vie moyenne (t½) du zanubrutinib est d'environ 2 à 4 heures après une dose orale unique de 160 mg ou 320 mg de zanubrutinib. La moyenne géométrique (% CV) de la clairance orale apparente (CL/F) du zanubrutinib pendant la phase terminale était de 128 (61 %) l/heure. Après une dose unique de 320 mg de zanubrutinib radiomarqué administrée à des sujets en bonne santé, environ 87 % de la dose a été retrouvée dans les selles (38 % sous forme inchangée) et 8 % dans l'urine (moins de 1 % sous forme inchangée).
Populations particulières
Personnes âgées
L'âge (19 à 90 ans, âge moyen 65 ans ± 12,5) n'a eu aucun effet cliniquement significatif sur la pharmacocinétique du zanutrutinib d'après une analyse PK de population (N = 1 291).
Population pédiatrique
Aucune étude pharmacocinétique n'a été effectuée sur le zanubrutinib chez les patients âgés de moins de 18 ans.
Sexe
Le sexe (872 hommes et 419 femmes) n'a eu aucun effet cliniquement significatif sur la pharmacocinétique du zanutrutinib d'après une analyse PK de population.
Origine ethnique
L'origine ethnique (964 Blancs, 237 Asiatiques, 30 Noirs et 25 classifiés comme autres) n'a eu aucun effet cliniquement significatif sur la pharmacocinétique du zanutrutinib d'après une analyse PK de population.
Poids corporel
Le poids corporel (36 à 149 kg, poids moyen 76,5 kg ± 16,9) n'a eu aucun effet cliniquement significatif sur la pharmacocinétique du zanutrutinib d'après une analyse PK de population (N = 1 291).
Insuffisance rénale
Le zanubrutinib fait l'objet d'une élimination rénale minime. D'après l'analyse PK de population, l'insuffisance rénale légère ou modérée (clairance de la créatinine [ClCr] ≥ 30 ml/min, estimée par l'équation de Cockcroft-Gault) n'a eu aucune influence sur l'exposition au zanubrutinib. L'analyse a été basée sur 362 patients ayant une fonction rénale normale, 523 patients ayant une insuffisance rénale légère, 303 ayant une insuffisance rénale modérée, 11 ayant une insuffisance rénale sévère, et un patient avec une IRCT. Les effets de l'insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) et de la dialyse sur la pharmacocinétique du zanutrutinib est inconnu.
Insuffisance hépatique
L'ASC totale du zanubrutinib a augmenté de 11 % chez les patients atteints d'insuffisance hépatique légère (classe A de Child-Pugh), de 21 % chez les patients atteints d'insuffisance hépatique modérée (classe B de Child-Pugh) et de 60 % chez les patients atteints d'insuffisance hépatique sévère (classe C de Child-Pugh) par rapport aux patients ayant une fonction hépatique normale. L'ASC non liée du zanubrutinib a augmenté de 23 % chez les patients atteints d'insuffisance hépatique légère (classe A de Child-Pugh), de 43 % chez les patients atteints d'insuffisance hépatique modérée (classe B de Child-Pugh) et de 194 % chez les patients atteints d'insuffisance hépatique sévère (classe C de Child- Pugh) par rapport aux patients ayant une fonction hépatique normale. Une corrélation significative a été observée entre le score Child-Pugh, lasérum-albumine à la référence, la bilirubine sérique à la référence et le temps de prothrombine à la référence, et l'ASC du zanubrutinib non lié.
Études in vitro
Enzymes CYP
Le zanubrutinib est un inducteur faible du CYP2B6 et du CYP2C8. Le zanubrutinib n'est pas un inducteur du CYP1A2.
Co-administration avec les substrats/inhibiteurs de transport
Le zanubrutinib est vraisemblablement un substrat de la P-gp. Le zanubrutinib n'est ni un substrat ni un inhibiteur de OAT1, OAT3, OCT2, OATP1B1 ou OAIP1B3.
Interactions pharmacodynamiques
Une étude in vitro a
montré que l'interaction pharmacodynamique potentielle entre le
zanubrutinib et le rituximab est faible et que le zanubrutinib est peu
susceptible d'interférer avec l'effet ADCC induit par l'anticorps
anti-CD20.
Des études in vitro, ex vivo et chez l'animal ont montré que le
zanubrutinib n'a pas d'effet ou a un effet minimal sur l'activation des
plaquettes, l'expression des glycoprotéines et la formation de thrombus.
Brukinsa n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Une fatigue, des vertiges et une asthénie ont été rapportés chez certains patients traités par BRUKINSA ; ces effets doivent être pris en compte lors de l'évaluation de l'aptitude du patient à conduire des véhicules et à utiliser des machines.
Toxicité générale
Les profils de toxicité générale du zanubrutinib ont été caractérisés par voie orale chez des rats Sprague-Dawley pour un traitement de maximum 6 mois et chez des chiens beagle pour un traitement de maximum 9 mois.
Au cours d'études à doses répétées chez des rats sous traitement pendant une période allant jusqu'à 6 mois, une mortalité liée au produit expérimental a été relevée à la dose de 1 000 mg/kg/jour (81 x l'ASC clinique), associée à des découvertes histopathologiques au niveau du tractus gastrointestinal. D'autres découvertes ont été relevées essentiellement au niveau du pancréas (atrophie, fibroplasie, hémorragie et/ou infiltration de cellules inflammatoires) à des doses ≥ 30 mg/kg/jour (3 x l'ASC clinique), de la peau située autour du nez/de la bouche/des yeux (infiltration de cellules inflammatoires, érosion/ulcère) à partir de 300 mg/kg/jour (16 x l'ASC clinique) et du poumon (présence de macrophages dans les alvéoles) à une dose de 300 mg/kg/jour. Après un rétablissement de 6 semaines, toutes ces découvertes étaient complètement ou partiellement résolues, sauf les découvertes pancréatiques, jugées non pertinentes sur le plan clinique.
Au cours d'études à doses répétées chez des chiens sous traitement pendant une période allant jusqu'à 9 mois, des découvertes cliniques liées au produit expérimental ont été relevées essentiellement au niveau du tractus gastro-intestinal (selles molles/aqueuses/mucoïdes), de la peau (rash, décoloration rouge et épaississement/desquamation), des ganglions lymphatiques mésentériques, mandibulaires et intestinaux ainsi que de la rate (déplétion lymphoïde et érythrophagocytose) à des doses allant de 10 mg/kg/jour (3 x l'ASC clinique) à 100 mg/kg/jour (18 x l'ASC clinique). Après un rétablissement de 6 semaines, toutes ces découvertes étaient complètement ou partiellement résolues.
Carcinogénicité/génotoxicité
Aucune étude de carcinogénicité n'a été conduite avec le zanubrutinib.
Le zanubrutinib ne s'est pas révélé mutagène dans un essai de mutagénicité bactérienne (test d'Ames), ni clastogène dans un essai d'aberration chromosomique réalisé sur des cellules mammifères (cellules ovariennes de hamster chinois, CHO). Il ne s'est pas non plus révélé clastogène dans un test in vivo du micronoyau de la moelle osseuse chez le rat.
Toxicité sur la reproduction et le développement
Une étude combinée de fertilités masculine et féminine et de développement embryonnaire précoce a été menée chez des rats à des doses orales du zanubrutinib de 30, 100 et 300 mg/kg/jour. Aucun effet sur les fertilités masculine et féminine n'a été rapporté, mais la dose testée la plus élevée, des anomalies morphologiques de sperme et une augmentation de la perte après implantation ont été relevées. La dose de 100 mg/kg/jour est environ 13 fois plus élevée que l'exposition thérapeutique humaine.
Des études de toxicité sur le développement embryo-foetal ont été menées chez le rat et le lapin. Du zanubrutinib a été administré par voie orale à des rates gravides pendant la période de l'organogenèse à des doses de 30, 75 et 150 mg/kg/jour. Des malformations cardiaques (cœurs à 2 ou 3 cavités avec une incidence de 0,3 % à 1,5 %) ont été observées à tous les niveaux de dose en l'absence de toxicité maternelle. La dose de 30 mg/kg/jour est supérieure d'environ 5 fois à l'exposition thérapeutique humaine.
L'administration de zanubrutinib à des lapines gravides pendant la période de l'organogenèse à 30, 70 et 150 mg/kg/jour a entraîné une perte après implantation à la dose la plus élevée. La dose de 70 mg/kg est supérieure d'environ 25 fois à l'exposition thérapeutique humaine et est associée à une toxicité maternelle.
Dans une étude de toxicité sur le développement prénatal et postnatal, du zanubrutinib a été administré par voie orale à des rats à des doses de 30, 75 et 150 mg/kg/jour entre l'implantation et le sevrage. La progéniture des groupes à doses moyenne et élevée présentait un poids corporel réduit avant le sevrage, et tous les groupes de dose ont développé des anomalies oculaires (par exemple, cataracte, yeux saillants). La dose de 30 mg/kg/jour est supérieure d'environ 5 fois à l'exposition thérapeutique humaine.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Prescription hospitalière.
Prescription réservée aux médecins compétents en maladie du
sang.
Prescription réservée aux spécialistes et services
HEMATOLOGIE.
Remboursement en fonction de l'indication (JO du 12/01/2024) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- En monothérapie dans le traitement de patients adultes atteints de
lymphome de la zone marginale (LZM) ayant reçu au moins un traitement
antérieur à base d'anticorps anti-CD20 ;
- Traitement des patients adultes atteints d'une leucémie lymphoïde chronique (LLC) non précédemment traités, uniquement :
- chez les patients ne présentant pas de délétion
17p et/ou de mutation TP53 et inéligibles à un traitement à base de
fludarabine à pleine dose ;
- chez les patients présentant une délétion 17p et/ou une mutation TP53 ;
- Traitement des patients adultes atteints de LLC ayant reçu au moins un traitement antérieur ;
- En monothérapie dans le traitement de patients adultes atteints de
macroglobulinémie de Waldenström (MW) qui ont reçu au moins un
traitement antérieur.
Gélule
Gélule opaque blanche à blanc cassé de 22 mm de
longueur, portant la mention « ZANU 80 » à l'encre noire
Flacon en PEHD avec bouchon en polypropylène doté d'une sécurité enfant. Chaque flacon contient 120 gélules.
Chaque gélule contient 80 mg de zanubrutinib
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu d'une gélule
Cellulose microcristalline
Croscarmellose de sodium
Laurylsulfate de sodium (E487)
Silice, colloïdale anhydre
Stéarate de magnésium
Enveloppe des gélules
Gélatine
Dioxyde de titane (E171)
Encre d'impression
Résine gomme-laque (E904)
Oxyde de fer noir (E172)
Propylène glycol (E1520)